2021.01.15
Roles of genome-wide biased-allelic-expression effects in the development of liver hepatocellular carcinoma.
SOKENDAI Student Dispatch Program program year: 2019
Genetics 羅鈞軒
遺伝学コース
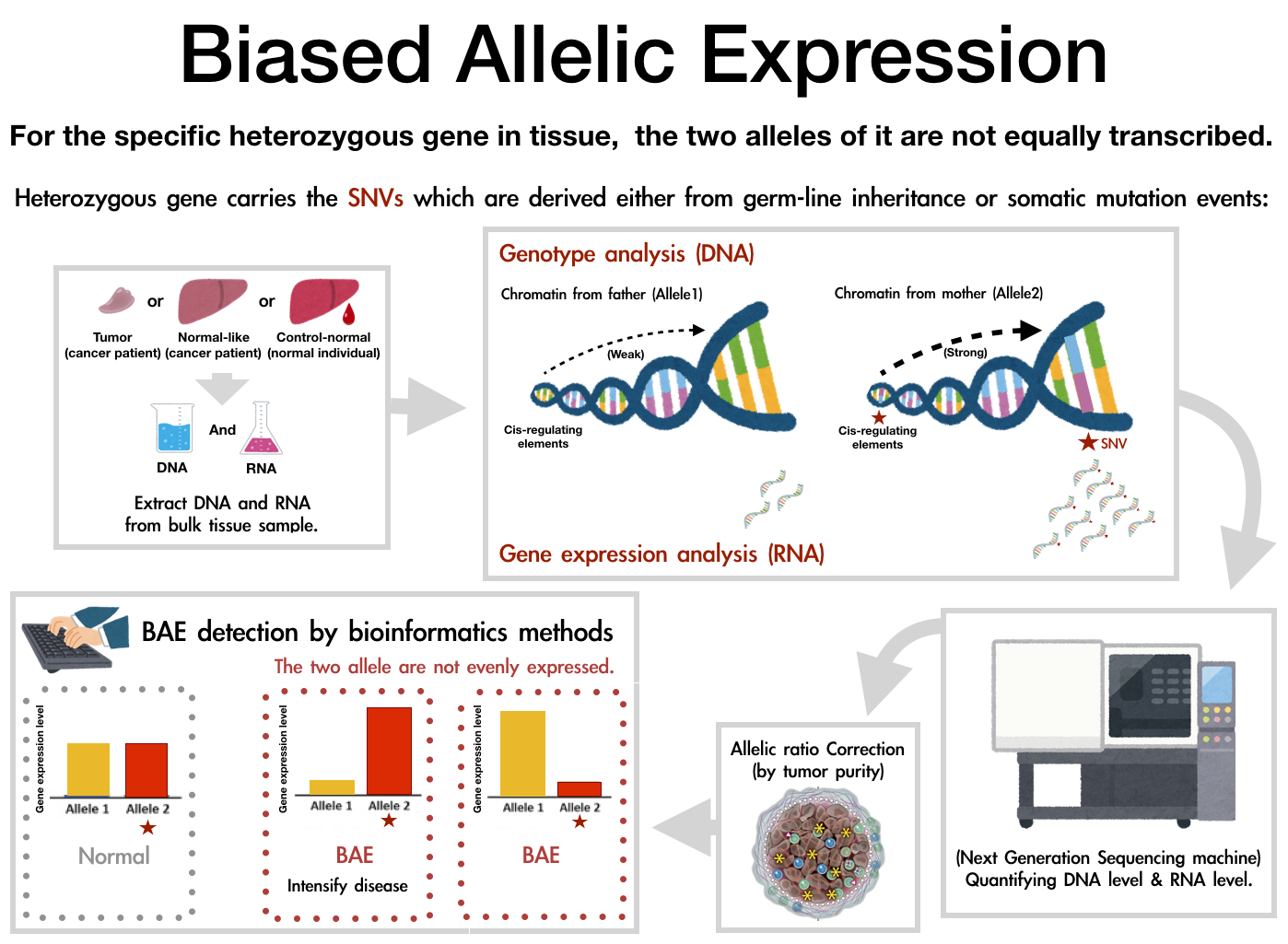
In diploid organisms including human, individuals have two copies (or alleles) for each gene on autosomal chromosomes, which are derived from mother and father respectively. If two alleles of a gene are not equally transcribed, we call the phenomenon as biased allelic expression (BAE). If there is any single nucleotide variant (SNV) in transcribed region of a gene at heterozygote state, we can quantify gene expression levels of the two alleles and evaluate whether allelic expressions are even or biased. There is a possibility that BAE effects are involved in the development of human diseases including cancers.
Background:
It has been reported that the changes in total expression levels of specific genes are related to cancer development. However, it has not been fully studied whether BAE effects can also be related to tumor development. I hypothesize that "the proportion of genes showing BAE effects" is much higher in the genes whose functions are related to tumor development than that in the other genes. Furthermore, I hypothesize that if a gene that is functionally relevant to cancer development is mutated, expression level of the mutated allele tends to be higher than that of wild-type allele.
Methods:
I focused on liver hepatocellular carcinoma (LIHC), the predominant form of liver cancer, for our BAE research. I integrated high-throughput sequencing data of DNA and RNA from two different online databases, which were named The Cancer Genome Atlas (TCGA) project and Genotype-Tissue Expression (GTEx) project. The TCGA enables us to compare each liver tumor tissue to its adjacent normal-like tissue from the same LIHC patient. The GTEx database provides us sequencing data for normal liver tissues from non-cancer individuals.
Result:
The result of this study suggests that differences in gene expression levels between LIHC and adjacent normal liver tissues were caused by genomic instability, which was indicated by higher frequency of DNA segmental insertion/deletion events.
I analyzed BAE effects in a genome-wide scale by focusing on those heterozygous genes with germ-line SNVs, which were directly inherited from their parents. As a result, I showed BAE effects were much more prevalent in LIHC tumors than in tumor-adjacent and normal liver tissues. Particularly, BAE effects in major histocompatibility complex (MHC) pathway related genes were significantly higher in LIHC tumors. It is suggested that stronger BAE effects in MHC genes in LIHC tumors are related to the escape of cancer cells by immunoediting process, which leads to the immune cells to fail the recognition of cancer cells. I detected tumor-specific BAE effects in genes that are presumably associated with the survival or selection of pro-tumor cells inside body.
Besides, I analyzed BAE effects of those heterozygous genes with somatically mutated SNVs, which were genetic alterations occurred in tumors. Remarkably, mutations on tumor protein p53 (TP53) most frequently represented BAE effects, which were accompanied by higher expressional levels of the mutated allele.
Conference Name
American Society of Human Genetics (ASHG) Annual Meeting 2019
Poster title
Roles of genome-wide biased-allelic-expression (BAE) effect in tumor development from the perspective of liver hepatocellular carcinoma (LIHC).
Period
Begin from 2019/10/11; End in 2019/10/19
Place
Houston, Texas / United States of America (USA)
Neoleukin Therapeutics, Inc.
Conference Contents
During the conference of ASHG 2019 Annual Meeting, the human geneticists from all over the world had come together for exchanging ideas and sharing new findings with other scientists. There are NCBI Hackathon and presentation sessions at HGSC, the chatting with other bioinformaticians helped me improve my analyzing protocols for utilizing on-line databases, and it also benefited my skills in bioinformatics and research competence in studying genetics.
Department of Genetics, Human Genetics Laboratory, LO, ChunHsuan (Jason)